Questões de Concurso
Para uerj
Foram encontradas 2.033 questões
Resolva questões gratuitamente!
Junte-se a mais de 4 milhões de concurseiros!
Q2518523
Medicina
Menina de 15 meses é encaminhada para avaliação de autismo e epilepsia. Apresentou desenvolvimento
normal até 1 ano de idade, chegando a falar algumas palavras, bater palmas e interagir bem. Desde então, a
família vem percebendo que a menor não progrediu o vocabulário e não se interessa por interagir com outras
crianças. Também está apresentando frequentes crises convulsivas. Os professores relatam intensas
estereotipias das mãos, principalmente em linha média, com dificuldades para as atividades. Para corroborar
a principal hipótese diagnóstica, deve ser buscada, no exame físico desse caso, a ocorrência de:
Q2518522
Medicina
Lactente de 4 meses é encaminhada ao ambulatório de Genética por atraso do desenvolvimento
neuropsicomotor (DNPM), fenda palatina ampla, cardiopatia congênita (estenose pulmonar), cisto renal
direito, hipotriquia e hipoplasia de unhas, dismorfias faciais (telecanto, nariz muito pequeno, orelhas pequenas
e displásicas, microglossia, micrognatia), web neck e braquidactilia. É a primeira filha de pais não
consanguíneos. A mãe teve diabetes gestacional com uso de insulina. A criança nasceu adequada para a
idade gestacional. Ao exame físico atual, verificam-se peso = 6,3kg (44%), comprimento = 62cm (48%) e
perímetro cefálico = 40,5cm (47%); dermatite seborreica e sulcos cutâneos transversais em braços, coxas e
tronco; linfedema leve em região dorsal dos pés; genitália externa feminina com grandes lábios proeminentes,
pigmentados e clítoris alongado. Os exames revelam sorologia TORCHS IgM: negativa; USG-Transfontanela:
normal; avaliação oftalmológica: coloboma de retina bilateral. Os testes genéticos indicam cariótipo 46,XX [20]
e Array-CGH arr(1-22, X)x2. Sequenciamento completo do exoma indica: (1) variante patogênica (c.1904del;
p.Lys635Argfs*10) no gene LIG4 (Ligase IV, DNA, ATP-dependent) em heterozigose, associada à síndrome
LIG4 (imunodeficiência combinada grave com fotossensibilidade, telangiectasia, psoríase, instabilidade
cromossômica, pancitopenia e atraso no desenvolvimento e crescimento), autossômica recessiva; (2) variante
provavelmente patogênica (c.500T>C; p.Leu167Pro) no gene PIGL (Phosphatidylinositol Glycan Anchor
Biosynthesis Class L Protein), em heterozigose, associada à síndrome CHIME (coloboma, congenital heart
disease, ichthyosiform dermatoses, impaired intellectual development, and ear anomalies syndrome),
autossômica recessiva. Nesse caso, a melhor interpretação e a conduta, respectivamente, são:
Q2518521
Medicina
Lactente de 2 meses é encaminhada para avaliação genética por aniridia bilateral e discreta opacidade
do cristalino em olho direito, cardiopatia congênita (estenose pulmonar moderada e FOP), grave atraso do
desenvolvimento neuropsicomotor (DNPM), hipotonia intensa, alterações da genitália (grandes e pequenos
lábios espessos e pigmentados) e dismorfias faciais (assimetria, ptose palpebral, telecanto, nariz pequeno,
microstomia e micrognatia). É a primeira filha de pais jovens, não consanguíneos. A gestação e o nascimento
não tiveram intercorrências. Ao exame físico, verificam-se peso = 5,6kg (75%), comprimento = 58cm (68%) e
perímetro cefálico = 38,5cm (58%). Apresenta infecção do trato urinário de repetição e doença do refluxo
gastroesofágico. A sorologia TORCHS está negativa e o estudo de imagem do cérebro e abdômen, normal.
Quanto à avaliação genética, apresenta: cariótipo 46,XX [30] e Array-CGH arr(1-22, X)x2. O sequenciamento
completo do exoma identificou variante provavelmente patogênica em heterozigose (c.6809-1G>C – posição
canônica de sítio de splicing) (p.?) no gene ITPR1 (Inositol 1,4,5-Triphosphate Receptor, Type 1), que está
associado à síndrome de Gillespie (aniridia, ataxia cerebelar, hipotonia intensa, atraso do DNPM e deficiência
intelectual) e às ataxias espinocerebelares 15 e 29 (ataxia cerebelar congênita não progressiva). Nesse caso,
considerando a variante como provavelmente patogênica, o diagnóstico mais adequado e a correspondente
interpretação, respectivamente, são:
Q2518520
Medicina
Adolescente de 16 anos, encaminhado ao ambulatório de genética para avaliar displasia esquelética,
é filho de pais jovens, saudáveis e não consanguíneos. Há relato de “artrite” com dor articular crônica
desde a primeira infância, cujo acompanhamento na reumatologia excluiu diagnóstico de doenças
autoimunes. Foi realizada cirurgia para correção de fenda palatina no primeiro ano, que evoluiu com
episódios recorrentes de otite média e atraso nos marcos do desenvolvimento motor (deambulou aos 3
anos), mas com cognitivo adequado à idade. Ao exame físico atual, verificam-se estatura = 122cm e
perímetro cefálico = 53,5cm; rizomelia, tórax curto, cifoescoliose, pelve estreita, pescoço curto,
hipopolasia de face média, ponte nasal plana e globos oculares proeminentes; articulações com rigidez e
aumento de volume. A audiometria revela hipoacusia condutiva e a avaliação oftalmológica indica miopia
severa. A dosagem de glicosaminoglicanos urinários mostra aumento leve de sulfato de queratan. O
estudo radiológico verifica vértebras com fendas coronais, fêmures em forma de haltere, megaepífises e
matriz interterritorial com aparência de “queijo suíço”. O caso descrito corresponde a um paciente com:
Q2518519
Medicina
As displasias ósseas constituem um grupo heterogêneo de doenças caracterizadas por alteração
primária do tecido ósseo e/ou cartilaginoso. O fenótipo em geral é caracterizado por baixa estatura
desproporcional (nanismo rizomélico) e desenvolvimento cognitivo preservado. O padrão de herança é,
primordialmente, autossômico dominante. A osteocondrodisplasia com padrão de herança autossômico
recessivo, na qual o desenvolvimento cognitivo NÃO é preservado, é a:
Q2518518
Medicina
Recém-nascida de parto cesáreo na 39ª semana de gestação é a primeira filha de pais não
consanguíneos saudáveis. A mãe tem 36 anos e o pai, 55. Um exame ultrassonográfico na 26ª semana
mostrou macrocefalia e rizomelia fetal. Ao nascimento, avaliou-se peso = 2,9kg, comprimento = 44cm e
perímetro cefálico = 36cm. Ao exame físico, verificaram-se hipotonia, macrocefalia, fronte ampla e
proeminente, hipoplasia de face média, rizomelia e braquidactilia. O raio X panorâmico identificou corpos
vertebrais pequenos com estreitamento caudal interpedicular progressivo, asas ilíacas pequenas e
estreitamento da incisura sacrociática, além de ossos tubulares curtos com alargamento metafisário.
Diante dessa situação, o exame complementar a ser solicitado é:
Q2518514
Medicina
Texto associado
Considere o heredograma a seguir, que representa uma doença com padrão de herança aotossômico dominante, para responder à questão.
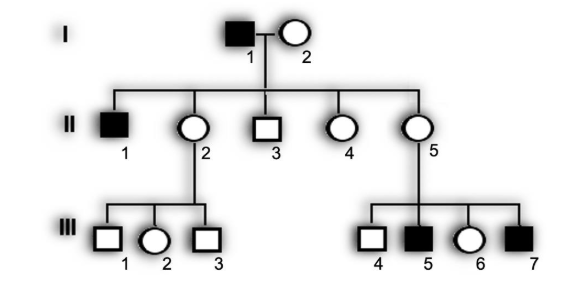
A condição genética representada pelo heredograma é:
Q2518513
Medicina
Texto associado
Considere o heredograma a seguir, que representa uma doença com padrão de herança aotossômico dominante, para responder à questão.
O heredograma representa que a:
Q2518512
Medicina
O gene Short Stature Homeobox (SHOX), localizado na região pseudoautossômica dos
cromossomos X (SHOX) e Y (SHOXY), codifica uma proteína que faz parte de uma grande família de
genes homeobox, um fator de transcrição, que atua durante o desenvolvimento embrionário inicial,
particularmente importante no crescimento e maturação dos ossos dos membros. A haploinsuficiência do
gene SHOX está relacionada a várias condições genéticas que cursam com déficit de crescimento. Uma
condição genética causada por haploinsuficiência do gene SHOX é a:
Q2518510
Medicina
A síndrome de Feingold tipo 1 (FGLDS1) é caracterizada por combinações variáveis de microcefalia,
malformações de membros, atresias de esôfago e duodeno, baixa estatura, atraso do desenvolvimento
neuropsicomotor e deficiência intelectual. É causada por mutações patogênicas em heterozigose no gene
MYCN (MYCN proto-oncogene, bHLH Transcription Factor). A síndrome de Feingold tipo 2 (FGLDS2)
tem fenótipo semelhante à FGLDS1 e é causada por:
Q2518509
Medicina
Os microRNAs (miRNA) são uma classe específica de genes de RNAnc com cerca de apenas 22
bases de comprimento. A função dos miRNA em relação ao genoma é regular:
Ano: 2024
Banca:
UERJ
Órgão:
UERJ
Prova:
UERJ - 2024 - UERJ - Médico - Endocrinologia - Diabetes e Metabologia |
Q2518505
Medicina
Em pacientes com DM2 assintomáticos em risco de progressão para insuficiência cardíaca, a
estratégia inicial recomendada para identificação precoce desses pacientes é:
Ano: 2024
Banca:
UERJ
Órgão:
UERJ
Prova:
UERJ - 2024 - UERJ - Médico - Endocrinologia - Diabetes e Metabologia |
Q2518503
Medicina
A classe de medicamentos que pode ser considerada para a redução da esteatose em adultos com
DM, sobrepeso/obesidade e doença hepática esteatótica associada à disfunção metabólica (MASLD) é:
Ano: 2024
Banca:
UERJ
Órgão:
UERJ
Prova:
UERJ - 2024 - UERJ - Médico - Endocrinologia - Diabetes e Metabologia |
Q2518502
Medicina
O critério diagnóstico de DMG é a:
Ano: 2024
Banca:
UERJ
Órgão:
UERJ
Prova:
UERJ - 2024 - UERJ - Médico - Endocrinologia - Diabetes e Metabologia |
Q2518501
Medicina
A metformina continua sendo um dos fármacos de primeira linha para tratamento do DM2. Na sua
prescrição, é necessário o cuidado com a:
Ano: 2024
Banca:
UERJ
Órgão:
UERJ
Prova:
UERJ - 2024 - UERJ - Médico - Endocrinologia - Diabetes e Metabologia |
Q2518499
Medicina
O fator de risco adicional para o aparecimento de diabetes em indivíduos com sobrepeso e obesidade é:
Ano: 2024
Banca:
UERJ
Órgão:
UERJ
Prova:
UERJ - 2024 - UERJ - Médico - Endocrinologia - Diabetes e Metabologia |
Q2518497
Medicina
A ordem correta da resposta hormonal, da mais precoce para a mais tardia, na presença de
hipoglicemia em pessoas sem diabetes é:
Ano: 2024
Banca:
UERJ
Órgão:
UERJ
Prova:
UERJ - 2024 - UERJ - Médico - Endocrinologia - Diabetes e Metabologia |
Q2518493
Medicina
Uma jovem assintomática, aos 14 anos, com IMC = 21kg/m2
, possuía resultado de exame laboratorial de
rotina glicemia de jejum (GJ) = 131mg/dL. Aos 19 anos, foi solicitada nova GJ, cujo resultado foi de 139mg/dL
com hemoglobina glicada = 6,6%, anti-GAD < 5 (VR- até 5UI/mL), anti-IA2 = 3 (VR até 10UI/mL) e peptídeo C =
3,7ng/dL. Atualmente, permanece assintomática. Há história familiar de “glicose alterada” para a mãe, além de
avó e bisavó maternas. O tipo mais provável de diabetes, para esse caso, é:
Ano: 2024
Banca:
UERJ
Órgão:
UERJ
Prova:
UERJ - 2024 - UERJ - Médico - Endocrinologia - Diabetes e Metabologia |
Q2518492
Medicina
Em um exame laboratorial cujo resultado corresponde ao diagnóstico de diabetes, os valores de
hemoglobina glicada (em %), glicemia de jejum (em mg/dL) e glicemia duas horas após a sobrecarga com
75g de glicose anidra, durante o teste de tolerância oral à glicose (em mg/dL), respectivamente, serão de:
Ano: 2024
Banca:
UERJ
Órgão:
UERJ
Prova:
UERJ - 2024 - UERJ - Médico - Endocrinologia - Diabetes e Metabologia |
Q2518491
Medicina
As metas de hemoglobina glicada recomendadas para idosos com DM saudáveis, frágeis e muito
comprometidos, respectivamente, são: